|
Guidelines at Diagnosis | About Clinical Trials
|
|
|
evidence-based support and information |
|
|
|
|
Study questions - relevance to patient needs and goals?
Randomized trials - common issues:
Justice (Belmont principle) - patient selection / eligibility:
Respect for persons (Belmont principle): Feasibility - is study protocol attractive to patients?
Do no harm - (beneficence)
|
|
Essential elements of Informed Consent fda.gov |
|||||||
|
Illustrations should be provided to present complex information |
|||||||
|
Day calendar showing what they patients can expect
|
Sources of Bias and Science-based Methods to Minimize
|
Abbreviations Oncology-Research-Glossary-10-30-15-NCI.pdf |
|||||||||
PAL items
|
|||||||||
|
End Points and United States Food and Drug Administration |
|||||||||
|
Design of Phase I Combination Trials: |
|||||||||
Dose Escalation Methods in Phase I Cancer Clinical Trials
|
|||||||||
|
Eliminating Bias in Randomized Controlled Trials: |
|||||||||
|
Bias in randomized controlled trials blackwellpublishing.pdf |
|||||||||
|
NCTN 2014 Working Group Report on Cooperative Group Trials |
|||||||||
|
NEJM - Perspective: Expediting Drug Development — |
|||||||||
|
FDA Patient Representative Workshop - videos available for each session http://bit.ly/YXBAuS |
|||||||||
|
What is advocacy? wpas-rights.org All people develop attitudes, preferences and biases. However, in order to be an effective advocate, you must be able to recognize your own attitudes, preferences, and biases. If you don't your attitudes are likely to interfere with your judgment. You could take positions which reflect your biases, rather than the choices of the people for whom you advocate. You may interpret the actions of others cynically, or naively, and thereby lose your ability to work effectively for the interests of others. |
Informed Consent and Concept Review Materials
|
CTEP forms for advocates and investigators CTEP |
|
|
Tables of Possible Side Effects for Commonly-Used Oncology Drugs |
|
|
Informed Consent Template for Adult Cancer Trials Cancer.gov |
|
|
Simplification of Informed Consent Documents Cancer.gov |
|
|
Consent, at-a-Glance, compiled by PAL discussion.pdf |
Advocacy Programs
|
Advocacy Programs (CARRA, FDA Patient consultants programs, NCI Director's Liaison, IRB, etc.)
|
|||
|
Clinical Trial Design and Participation Consulting with Patients, the FDA, Drug Sponsors, the NCI, and community physicians. Establishing credibility through hard work and financial independence. |
|||
|
Overcoming obstacles to participation in clinical trials Patient Perspective |
Biospecimen issues and resources
|
Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): Explanation and Elaboration http://1.usa.gov/1wb5zim |
|
|
JCO, 2012: Ethics of Mandatory Research Biopsy for Correlative End Points Within Clinical Trials in Oncology http://bit.ly/1s0u1vx |
|
|
2006 by American Society of Clinical Oncology |
|
|
Issues in collecting, processing and storing human tissues and associated information to support biomedical research http://1.usa.gov/13XxrJr |
|
|
NCI Biospecimen Best Practice - Consent, ownership, standards http://biospecimens.cancer.gov/bestpractices/2011-NCIBestPractices.pdf |
Primers on clinical research
|
CISN.org: primer on research http://cisncancer.org/research/index.html |
|
|
Introduction to Pharmacokinetics and Pharmacodynamics ashp.org |
|
|
An advocate's notes on dose-finding studies Dose - the importance of getting it right |
Terminology
|
Glossary of lay terms for side effects http://humansubjects.stanford.edu/new/docs/glossary_definitions/lay_language.pdf |
|
|
Clinical trial terminology and acronyms http://humansubjects.stanford.edu/research/documents/clinical_trial_terms.acronyms_AID01020.AID03022.pdf |
|
Institutional Review Board Member Handbook |
|
|
Principles of Clinical research based on the Belmont Report |
• The principle of respect for persons includes two separate moral requirements:
1) the requirement to acknowledge autonomy and 2_ the requirement to protect those with diminished autonomy” (The Belmont Report)
In practice, this means that individuals have a right to decide for themselves whether to participate in research. You may not use information about people without first getting their informed consent. Special care must be taken with people who are unable to understand or who are particularly susceptible to coercion.
• Beneficence
Two general rules apply:
(1) do not harm and
(2) maximize possible benefits and minimize possible harms.” (The Belmont Report)
In practice, this means that it is not OK to use people for research unless the research is likely to have some benefit. Furthermore, this benefit must outweigh the risks.
• Justice
Justice requires that people be treated fairly. Researchers should not take from research participants without giving back:
“For example, the selection of research subjects needs to be scrutinized in order to determine whether some classes (e.g., welfare patients, particular racial and ethnic minorities, or persons confined to institutions) are being systematically selected simply because of their easy availability, their compromised position, or their manipulability, rather than for reasons directly related to the problem being studied.
Whenever research supported by public funds leads to the development of therapeutic devices and procedures, justice demands both that these not provide advantages only to those who can afford them and that such research should not unduly involve persons from groups unlikely to be among the beneficiaries of subsequent applications of the research.”
"For many centuries doctors used leeches and lancets to relieve patients of their blood. They KNEW bloodletting worked. EVERYBODY said it did. When you had a fever and the doctor bled you, you got better. EVERYONE knew of a friend or relative who had been at death’s door until bloodletting cured him. Doctors could recount thousands of successful cases."
See also: When Laypersons Give Medical Advice
The lesson from history described above teaches us that the clinical observations of medical professionals cannot be relied upon when judging the efficacy of treatments. Scientific methods are needed ... and have been the foundation of modern medicine.
The most common purpose of clinical trials is to test a study protocol in order to judge if it will provide meaningful clinical benefit - compared to the natural course of the disease or the disease treated differently. Clinical benefit from a treatment is defined as an improvement in survival or quality of life. It can be very challenging to demonstrate that a new treatment is an improvement over existing approaches. For example the increase in response rate might be offset by impacts on quality of life or late toxicities. Comparing survival is the most objective way to compare outcomes ... but this requires large studies to account for differences in patient and disease characteristics - and, particularly for the indolent lymphomas, the influence of the tested treatments on response to subsequent therapies.
Background for new advocates:
Individual outcomes (even validated case reports) cannot be used as evidence of clinical benefit for the patient or others - because we cannot know how an individual would have done had they approached it differently - or if that patient's disease or risk factors are very much like other patients.
So to judge how likely a new treatment will help others often requires controlled trials on large groups of patients with the same diagnosis, and similar risk factors. The most objective evidence of how treatments compare will come from large studies that randomly assign patients to the different treatments to assure that the risk factors are balanced in the compared groups.
Improved methods for evaluating the underlying biology of lymphoma will reveal new subtypes that were formerly considered one disease (such as Germinal B-Cell versus Activated B-Cell Diffuse Large B-cell Lymphoma). So we can expect that patient selection for trials will increasingly be based on new tests that identify these subtypes -- instead of doing "all-comer" trials and that the increasing number of subtypes will increase the challenge of carrying out sufficiently powered clinical trials.
See also The Problems with Testimonials AND Evaluating medical claims and dataDemonstrating in controlled clinical trials that a treatment improves overall survival is the most direct evidence that the treatment provides meaningful clinical benefit - as a difference in overall survival accounts for all effects of the treatments - good and bad, measured and not measured. When two or more treatments provide the same or unknown survival benefit then the costs, and anticipated impacts on quality of life can determine which is superior.
... However, there is a major issues with the survival endpoint, as it's not ethical to wait and see which approach to treatment improves the survival of two groups of patients. Other treatments will be given to the patients at relapse, which confounds assessment of the survival endpoint. So we need other ways to judge if a therapy reasonably predicts improved survival. These measures are called surrogate endpoints.
Surrogate endpoints are like the parts of the elephant touched by the six blind men of Indostan - each sensation reveals a different aspect of the elephant - the toe, the ear, the trunk, but not the whole picture.
The surrogate endpoints commonly used by investigators to measure outcomes in clinical trials are: tumor response rates, relapse rates, progression events, side effects rates, and the incidence of death. The events are counted and plotted over time to see how often they occur and when - relative to the beginning or the end of treatment - or relative to the dose of a study drug in phase I testing.
The purpose of measuring endpoints is to compare the effects of the study drug - good and bad, on the patient or the disease. Quality of life endpoints measure effects on the person - reported by the persons. Disease event endpoints measure effects on the disease - reported by imaging.
EFFICACY ENDPOINTS
Must be weighed against the toxicities and risks of the intervention,
compared to the risk of the disease untreated or treated differently.
These terms are often used to evaluate and compare responses to therapies (outcomes) in clinical studies. They do not measure or predict individual outcomes, but are used to show how effective the treatment was for a given group of patients in a specific time interval.
Actuarial Survival or Overall Survival (OS): A measures of the number of patient who are still alive following treatment in a fixed time interval.Event free survival (EFS): A measure of the number of patients who are still alive and have not had a relapse (the event) following treatment in a fixed time interval. For example: After 84 month the actuarial survival rate was 93.1% and event-free survival rate was 87.3% for a given therapy.
Time to Progression
(TTP) is a measure used to estimate the response to treatment, generally in a clinical trial setting. As such it is often called an endpoint. It goes something like this:You get a baseline scan, usually CT, before treatment to measure tumor burden. A second CT after treatment measures the response, generally as a % of decrease in tumor burden (50% or greater being a Partial Response, and a CR indicating a Complete Response.)
From that point the time interval between the response to treatment and the time the disease starts to show evidence of growing (or recurring if a CR), is Time To Progression. Of course how often CTs are taken can affect this measure greatly, so TTP can only be an estimate.
Progression free survival (PFS)
measures time to progression, relapse, and to death from any cause. The rationale for including "death from any cause" - even if not apparently related to the disease or treatment - is that it's not always possible to know what's related; and accidental or unrelated causes will balance out when comparing groups treated with different protocols.Disease Free Interval
is similar to Time To Progression, but it differs in that it only relates to complete responses (CRs), because "free" indicates no disease measurable. So only a person who has had a CR can be among those measured for disease free interval; for partial responders this term does not apply.Patient reported outcomes (PRO)
captured the burden of the disease or treatments directly from the patient - outcomes that cannot be identified with standard tests, such as blood tests or imaging scans.
How much risk is acceptable for treatment (or in a clinical trial) can depend on the risk of the lymphoma and this can vary in the same diagnosis.
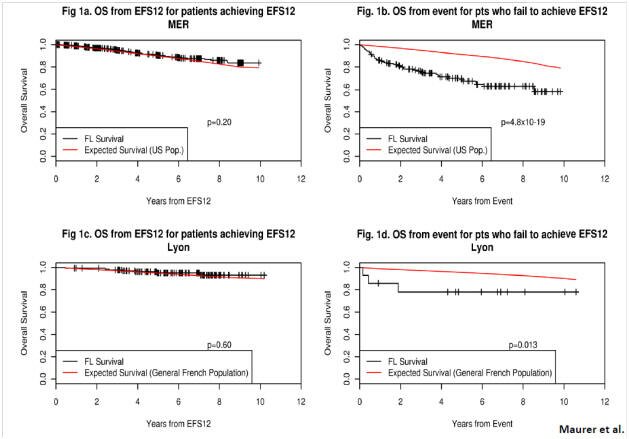
The following charts show different survival plots from time of progression after R-CHOP – showing clearly that early progression of disease (POD of less than 1 year) is worse in respect to OS than late POD). The red line in each chart shows the expected survival of the age-related normal population. The dark lines with hatches show FL patients.
Note that the two charts on the left show no difference from the expected survival of the normal population (red and dark lines overlap) when the Event Free Survival (EFS12) for FL patients after R-CHOP is 12 months or more.
Contrast this with the two charts on the right side, where the red and dark lines separate in patients who did not achieve EFS12.
The Symptom Management and Health-related Quality of Life (SxQoL) Steering Committee has produced a number of webinars to assist investigators in Concept Design and using PROs in clinical Trials. These webinars are listed below and are live on the NCI Division of Cancer Prevention Website: http://prevention.cancer.gov/news-events/events/videos/pro_symptommanagementwebinars.
Best Practices for Integrating Patient Reported Outcomes in Oncology Clinical Trials - The following webinar series is a collaboration of the International Society for Quality of Life Research and the National Cancer Institute. The videos in the series may be viewed independently or sequentially in the order listed below.
Session 1
David Cella, PhD presents
How to identify the PRO context for clinical trials and identify the relevant PRO domains
Air date: 09/04/2014
Session 2
Bryce B Reeve, PhD & Ethan M Basch, MD present
How to select the appropriate PRO measure
Air date: 09/03/2014
Session 3
Madeleine King, PhD, Michelle Naughton, PhD, & Lari Wenzel, PhD present
How to design a high quality study with PRO endpoints
Air date: 08/20/2014
Session 4
Amylou Dueck, PhD & Diane Fairclough, DrPH present
How to develop the statistical plan and sample size calculation for the PRO component of a clinical study
Air date: 08/19/2014
Session 5
Carol M Moinpour, PhD & Andrew Bottomley, PhD present
How to assure data quality for PROs in oncology clinical trials
Air date: 09/04/2014
Session 6
Michael Brundage MD & Melaine Calvert, PhD present
How to report PRO study findings from clinical trials
Air date: 08/19/2014
|
June 12: Dr. Sandra Mitchell, The key elements of the PRO-CTCAE measurement system are exhibited, and the research challenges, gaps in knowledge, and the issues that will need to be resolved to fully implement this new approach are examined. |